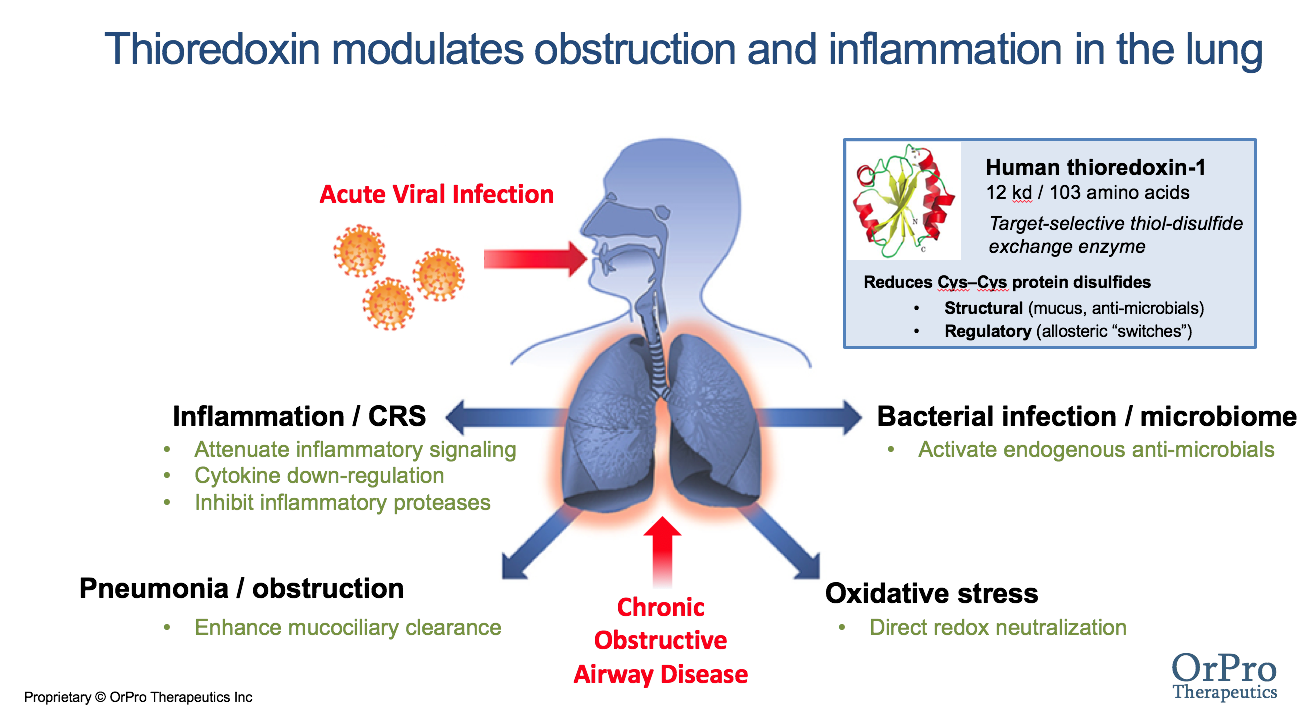
CF patients are frequently hospitalized for disease exacerbations and many ultimately require lung transplantation, which is life-extending but not curative. Until gene or protein-repair therapies are one day able to effect a true cure, disease-modifying strategies to prevent or slow progression to life-threatening symptoms are the best hope for patients today. Some patients carry CF alleles that can be targeted by small-molecule CFTR modulator drugs (Kalydeco®, Orkambi® Symdeko® and Trikafta®)that increase CFTR expression or restore partial ion channel activity. However, nearly 2000 unique mutations affecting CFTR have been identified, creating challenges for establishing genotype-independent modulator treatments. While modulator use may one day be extended to as many as 90% of CF patients even the best-responders still suffer declining lung function, with many patients responding variably or not at all. Consequently, CF standard-of-care remains largely symptomatic and directed against the later-stage manifestations of the disease – infection, inflammation, pancreatic insufficiency, and airway obstruction. In addition to CFTR modulators for those who are genetic candidates, the daily regimen for a CF patient typically includes inhaled antibiotics, anti-inflammatories, dietary enzyme supplementation, physical therapy, and inhaled mucolytic and airway hydration/cough inducing treatments. A large need therefore exists for new approaches that can reduce this treatment burden.